分子动力学(MD)模拟是探究生物分子机制的核心工具,其成功依赖于力场的准确性、效率和泛化能力。经典分子力场(MM)高效但精度受限,量子力学(QM)准确却计算开销巨大,机器学习力场(MLFF)作为桥梁应运而生。近年来,人工智能驱动的MD模拟快速发展,从静态结构预测向动态行为建模转型,为揭示信号通路、药物靶点结合等提供原子级洞见,推动生物机制阐明和药物发现。
2025年11月21日,清华大学生命科学学院王童课题组应邀在《结构生物学的当前观点》(Current Opinion in Structural Biology)杂志上发表题为“基于机器学习力场的AI驱动的生物分子模拟最新进展”(Recent advances in artificial intelligence–driven biomolecular dynamics simulations based on machine learning force fields)的综述文章。该文概述了MLFF的设计关键因素,总结了三大类MLFF的进展,分析了其局限性,并展望了其在全细胞多尺度模拟中的应用。
近年来,人工智能的兴起,如AlphaFold在蛋白质结构预测领域的突破,凸显了AI在计算结构生物学中的潜力。基于MLFF分子动力学模拟使用神经网络计算原子的受力并迭代更新位置和速度,能够追踪分子行为的时序演化,从而探查生物分子的复杂机制。
MLFF的成功取决于准确性、效率和泛化能力的平衡。准确性要求预测能量和力忠实再现量子势能面,以准确计算如蛋白折叠自由能等宏观性质。效率确保对于蛋白质折叠或变构调控的长时间尺度模拟,目前MLFF对典型生物体系在显式水环境中的模拟速度可达每天数纳秒。泛化能力则要求模型能够在有限数据的训练下,泛化到不同构象和分子上。基于片段化策略和MLFF的MD系统极大提高了泛化性,避免为每个系统从头训练的高成本。
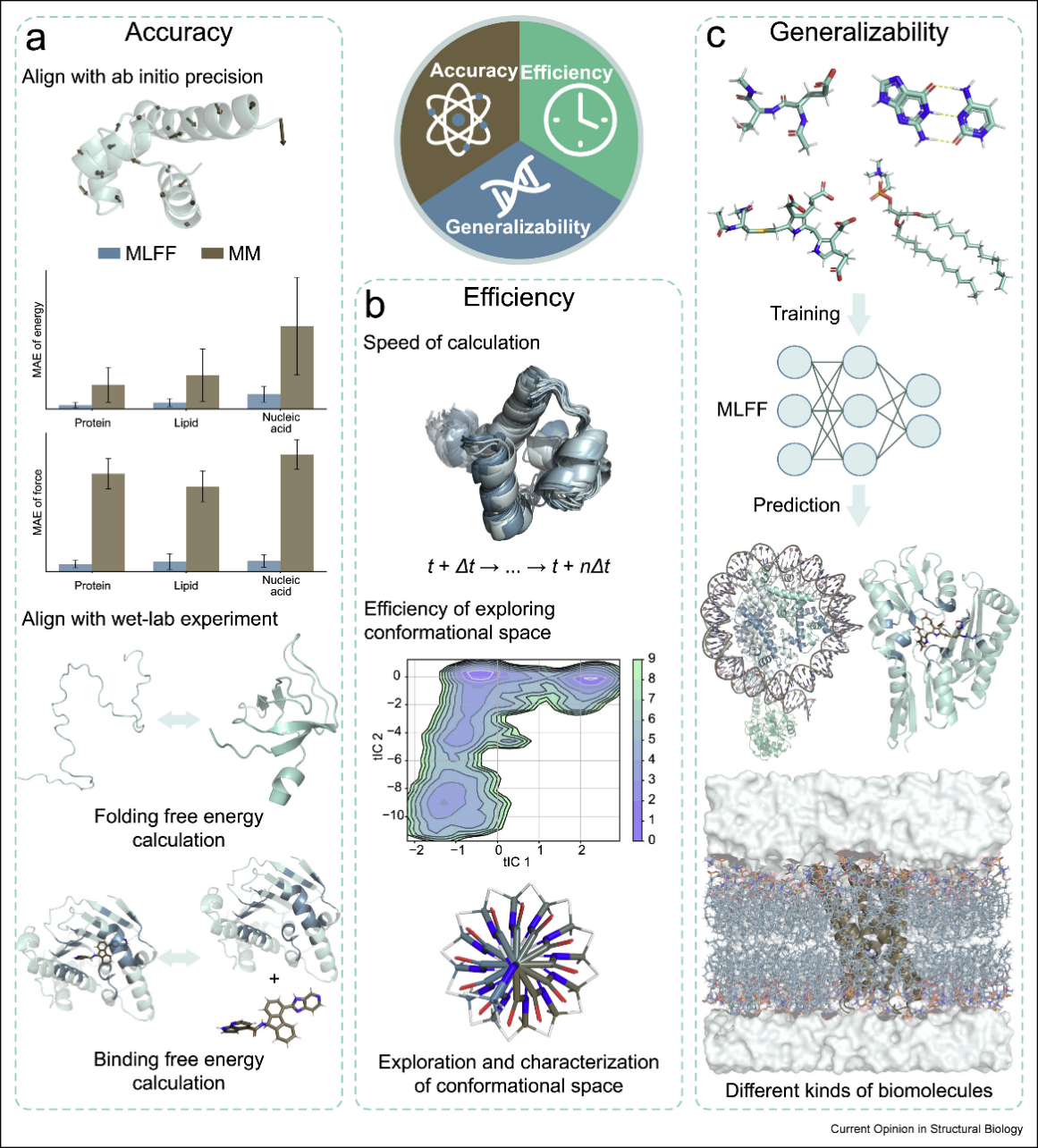
图1 机器学习力场设计中的关键因素:准确性、效率和泛化能力
通过神经网络参数化经典分子力场是MLFF的一大类别,如Espaloma, ByteFF等[1-2]。这些方法利用从头计算数据集优化经典力场的键合和非键合项参数,捕捉多体相互作用如极化和电荷转移,同时保留经典力场的计算效率。ByteFF在蛋白质和核酸模拟中表现出色,超越传统力场在能量和力计算的精度。
端到端训练的MLFF摒弃固定解析形式,直接建立原子坐标到势能面的映射,旨在突破经典力场限制以逼近从头算精度。相较于早期的不变性模型,引入等变图神经网络(EGNN)已成为主流,通过在网络中保留旋转和平移的几何对称性,显著提升了模型的数据利用率与准确性。以ViSNet为例,该模型采用高效的标量-矢量交互网络架构,并基于物理启发引入四体相互作用,在避免高昂计算成本的同时有效增强了对复杂几何特征及远程相互作用的捕捉,为生物大分子体系提供了兼具高精度与高效率的模拟方案[3]。
为克服端到端模型在泛化上的局限性,AI2BMD和GEMS等通用力场引入了片段化策略。AI2BMD基于二肽单元构建通用库,GEMS则融合bottom-up的小分子与top-down的特异性片段,实现了对大分子体系的高效扩展。实验表明,这些方法不仅能精确计算蛋白质热力学性质和折叠自由能,还能准确复现太赫兹光谱等动态特征,在万原子级溶剂化体系中实现了近从头算精度[4-5]。
尽管进展显著,MLFF仍面临准确性、效率与泛化性的多重挑战与内在权衡。局域截断限制了对长程静电相互作用的捕捉,计算效率仍显著慢于经典力场,制约了对慢速生物过程的探索。此外,各要素间难以兼得:提升模型精度往往以牺牲效率为代价,而追求广泛的化学空间覆盖则可能导致特定体系的预测精度下降。
展望未来,MLFF将与虚拟细胞模型和粗粒化表示整合,实现全细胞多尺度模拟,桥接计算预测与实验验证,加速生物机制阐明和治疗发现。
清华大学生命科学学院王童助理教授为本文通讯作者,王童课题组来自香港中文大学的崔涛镛实习生和来自北京大学的周雨涛实习生为本文共同第一作者。该工作得到了国家重点研发计划、清华-北大生命科学联合中心、北京生物结构前沿研究中心等项目的支持。
原文链接:https://doi.org/10.1016/j.sbi.2025.103191
参考文献:
[1] Takaba K, Friedman AJ, Cavender CE, Behara PK, Pulido I, Henry MM, MacDermott-Opeskin H, Iacovella CR, Nagle AM, Payne AM, Shirts MR. Machine-learned molecular mechanics force fields from large-scale quantum chemical data. Chemical science. 2024;15(32):12861-78.
[2] Zheng T, Wang A, Han X, Xia Y, Xu X, Zhan J, Liu Y, Chen Y, Wang Z, Wu X, Gong S. Data-driven parametrization of molecular mechanics force fields for expansive chemical space coverage. Chemical Science. 2025;16(6):2730-40.
[3] Wang Y, Wang T, Li S, He X, Li M, Wang Z, Zheng N, Shao B, Liu TY. Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing. Nature Communications. 2024 Jan 5;15(1):313.
[4] Wang T, He X, Li M, Li Y, Bi R, Wang Y, Cheng C, Shen X, Meng J, Zhang H, Liu H. Ab initio characterization of protein molecular dynamics with AI2BMD. Nature. 2024 Nov 28;635(8040):1019-27.
[5] Unke OT, Stöhr M, Ganscha S, Unterthiner T, Maennel H, Kashubin S, Ahlin D, Gastegger M, Medrano Sandonas L, Berryman JT, Tkatchenko A. Biomolecular dynamics with machine-learned quantum-mechanical force fields trained on diverse chemical fragments. Science Advances. 2024 Apr 5;10(14):eadn4397.

